
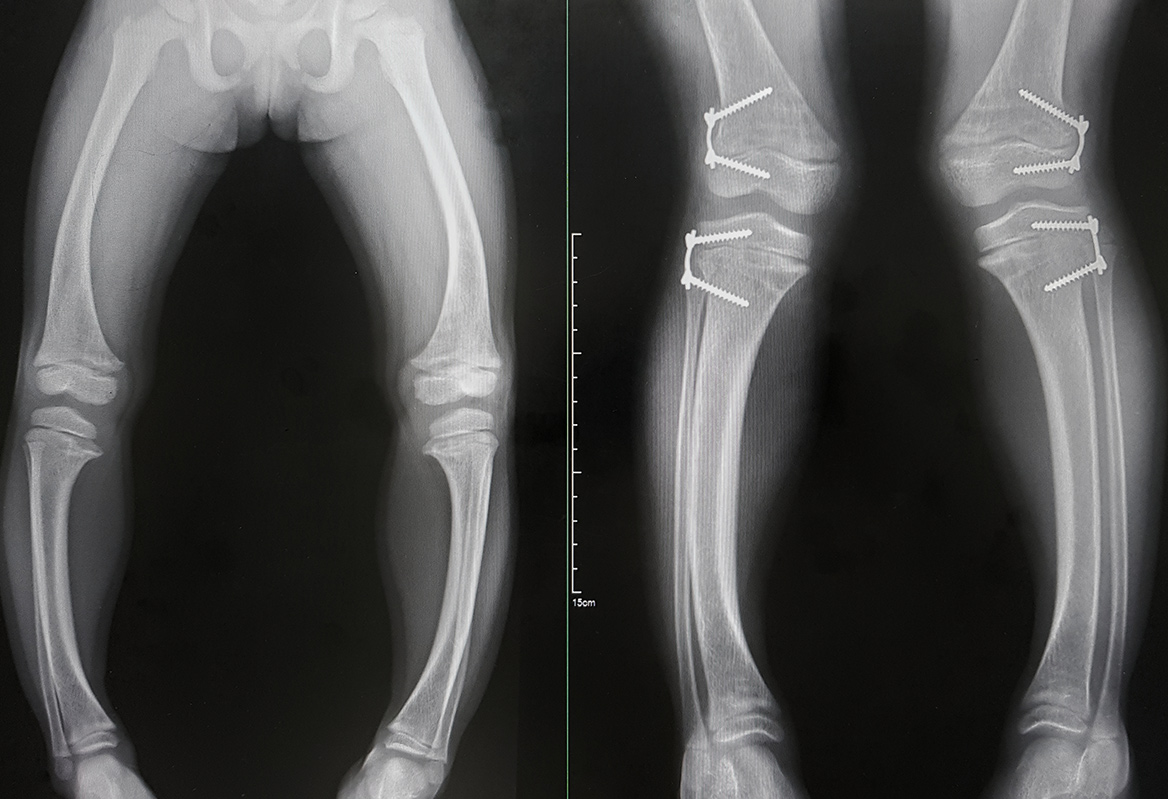
La hipofosfatemia ligada al cromosoma X (XLH) es una enfermedad caracterizada por la pérdida de fosfato a nivel renalI . Se considera una patología rara, además de progresiva y crónica, causada por la mutación del gen PHEX, ubicado en el locus Xp22 del cromosoma XI. Su prevalencia es de 1 por cada 20.000 personasII III, con mayor afectación en mujeresII III. Esta pérdida de fosfato afecta especialmente a la mineralización del hueso, dando lugar a raquitismo u osteomalacia, hipocrecimiento en el niño, fracturas y deformidades óseas, entre otrosI IV V VI. No obstante, la deficiencia de fosfato tiene una repercusión multisistémica, afectando a todo el organismoVI. Aunque suele manifestarse en los dos primeros años de vidaI, es una enfermedad de por vida que incluso puede detectarse con sintomatología, a veces aparentemente más leve, en la edad adultaVII.
Para hablar de esta patología, la compañía farmacéutica Kyowa Kirin ha celebrado la Reunión Nacional Exchange Hypophosfatemia Academy, en la que profesionales implicados en el manejo de la enfermedad han tratado su evolución y abordaje desde diferentes perspectivas, así como los avances terapéuticos para esta patología.
Detección precoz
Uno de los puntos destacados en esta reunión ha sido la importancia del diagnóstico temprano. Carmen de Lucas, del Servicio de Nefrología Infantil del Hospital Universitario Niño Jesús, comenta cómo, así, el tratamiento precoz puede mejorar la talla final del paciente y evitar muchas complicaciones. “Todos los médicos implicados en el seguimiento de los niños, el pediatra de atención primaria, traumatólogo, endocrinólogo o rehabilitador pueden ser los primeros en sospecharla clínicamente y hacer un estudio inicial. En ocasiones, el diagnóstico se retrasa debido a que, como un cierto grado de varo puede ser fisiológico en los niños, esta deformidad, a menudo, no se tiene en cuenta en pacientes menores de dos años, salvo que sea muy marcada”, comenta la doctora De Lucas.
Francisco Nieto, nefrólogo del Hospital Universitario de Málaga, señala lo determinante que es la mineralización ósea en la infancia, y cómo hay estudios que demuestran que el inicio tardío del tratamiento repercute negativamente sobre la talla final, las deformidades y el dolor. ”En los últimos años se han descrito formas de presentación atípicas que pueden dificultar el diagnóstico. Por eso es importante realizar estrategias que aumenten el conocimiento de la enfermedad sobre los profesionales de primera línea, ya que son cruciales para el diagnóstico precoz”, afirma el nefrólogo.

El desconocimiento de esta patología entre muchos de los profesionales, como algunos pediatras de atención primaria o de urgencias, puede generar en muchas ocasiones retrasos en el diagnóstico de estos pacientes, que idealmente se deberían detectar en los primeros dos años de vida. Además, Pedro Arango Sancho, miembro del servicio de Nefrología Pediátrica en el Hospital Sant Joan de Déu de Barcelona, considera que, aunque, no es infrecuente. “no siempre el desconocimiento es la base de este retraso diagnóstico, ya que la gran variabilidad fenotípica que presenta la enfermedad puede jugar un gran papel en el infradiagnóstico que observamos”.
Avances terapéuticos
Otro de los pilares tratados en la Reunión Nacional Exchange Hypophosfatemia Academy ha sido el desarrollo novedades terapéuticas. Así, los profesionales implicados en el manejo de esta oportunidad han tenido la oportunidad de compartir, de primera mano, su experiencia en este sentido.
“Llevamos 40 años usando un tratamiento que no actuaba contra el mecanismo fisiopatogénico de la misma y que además asociaba numerosos efectos adversos. Además, los resultados en muchos casos eran insuficientes. Todo esto, junto a una baja adherencia asociada, en parte, a la necesidad de múltiples tomas diarias (entre 3-5) de forma crónica”, afirma Pedro Arango.
Arango también destaca cómo recientes avances en el tratamiento pueden mejorar la calidad de vida de estos enfermos. “Hemos pasado de un tratamiento clásico con unas limitaciones y complicaciones específicas que esperábamos y controlábamos, a uno completamente nuevo que además de abrirnos nuevas esperanzas en el manejo de la patología nos aporta nuevos retos y dudas que creemos necesarios poner en común, generando con ello un foro de debate y crecimiento inigualable para todos”, comenta.
Francisco Nieto explica cómo esta nueva terapia consiste en “un anticuerpo monoclonal totalmente humano que inactiva el exceso de la hormona responsable de la enfermedad. Esto ha demostrado un beneficio no solo en la normalización de los niveles de fosfato en sangre, sino también una mejoría a nivel de las alteraciones radiológicas óseas, deformidades, dolor y calidad de vida”.
Hasta ahora el tratamiento se basaba en la administración crónica de sales de fósforo y suplementos de vitamina D activa. Carmen de Lucas indica cómo “actualmente contamos con un anticuerpo monoclonal humano contra el FGF23 que contrarresta sus efectos. Este tratamiento está autorizado en Europa para la XLH en niños y adolescentes de 1 a 17 años con signos radiográficos de enfermedad ósea, y en adultos. No obstante, en España se encuentra actualmente dentro de reembolso mientras el paciente tenga un esqueleto en crecimiento”, indica la doctora.
Principales retos
Respecto a los retos en el manejo óptimo de los niños con esta enfermedad, los expertos coinciden en seguir incidiendo en el diagnóstico y el tratamiento precoz.
“Es importante realizar campañas formativas a profesionales sanitarios, no solo a expertos en la enfermedad, sino también a aquellos profesionales que trabajan en primera línea, como médicos de atención primaria, urgencias, o, también, odontólogos. Debe mejorarse también el abordaje multidisciplinar, para lo cual se deben trabajar los nexos de comunicación entre las diferentes especialidades que atienden a estos pacientes. Además, es importante realizar la correcta transferencia a adultos, para lo que es esencial la comunicación entre especialidades pediátricas y de adultos”, afirma Francisco Nieto.
Carmen de Lucas insiste en “individualizar el tratamiento de cada paciente y tener una buena calidad asistencial, eficiencia y equidad”.
El objetivo en el manejo de los pacientes pediátricos con XLH clásicamente ha sido mejorar el raquitismo, normalizar o mejorar las malformaciones óseas asociadas, favorecer el crecimiento, reducir el dolor óseo y mejorar la salud dental. Para Pedro Arango “esto lleva asociado una mejoría en la movilidad y la calidad de vida de los pacientes. A pesar de ello, no suelen obtenerse con los tratamientos clásicos los objetivos de talla diana genética en estos pacientes, que en la mayoría de los casos es inferior a -2DE en ambos sexos. Además, el deterioro de la calidad de vida objetivado en estudios realizados mediante cuestionarios en los que se registran aspectos como el bienestar, la felicidad o la limitación funcional deben tenerse muy en cuenta para el desarrollo de estrategias y el diseño de mejorías en los tratamientos actuales”, concluye Arango.
REFERENCIAS:
I Haffner D, Emma F, Eastwood DM, et al. Consensus Statement. Evidence-based guideline. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019;15;435-455.
II Beck-Nielsen SS, et al. Incidence and prevalence of nutritional and hereditary rickets in southern Denmark Eur J Endocrinol 2009 Mar;160(3):491-497.
III Beck-Nielsen SS, et al. FGF23 and its role in X-linked hypophosphatemia-related morbidity Orphanet J Rare Dis. 2019;14(1):58.
IV Linglart A, Biosse-Duplan M, Briot K, et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect 2014; 3(1): R13-R30.
V Skrinar A, Dvorak-Ewell M, Evins A, et al. The lifelong impact of X-linked hypophosphatemia: Results from a burden of disease survey. J Endocr Soc. 2019;3:1321-1334.
VI Lambert AS, et al. X-linked hypophosphatemia: Management and treatment prospects. Joint Bone Spine. 2019;86(6):731-738.
VII Lo SH, Lachmann R, Williams A, et al. Exploring the burden of X-linked hypophosphatemia: a European multi-country qualitative study. Qual Life Res. 2020;29(7):1883-1893.
Este contenido ha sido desarrollado por UE Studio, firma creativa de branded content y marketing de contenidos de Unidad Editorial, para KYOWA KIRIN
On Ofrecido por KYOWA KIRIN Onvia Noticias de diariomedico.... https://ift.tt/3FCPbM4
No hay comentarios:
Publicar un comentario